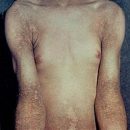
Primær immundefekt er medfødte overtrædelser af det immunsystem, der udvikler sig på grund af genetiske lidelser. Former af primær immundefekt er insufficensen i systemet med komplement, fagocytose, såvel som humoral og cellulær immunitet.
Indhold
Begrebet primær immundefekt
Primær immundefekt er medfødt svækkelse af immunsystemet, der er forbundet med genetiske defekter af en eller flere komponenter i immunsystemet, nemlig komplement, fagocytose, humoral og cellulær immunitet. Et fælles træk ved alle typer primære immundefekt er tilstedeværelsen af kroniske infektioner, der påvirker forskellige organer og væv og som regel forårsaget af den opportunistiske mikroflora. Opportunistiske infektioner er en stor gruppe af en række smitsomme sygdomme. Det kombinerer dem, at de kun forekommer med et udtalt fald i immuniteten. I en sund person eller en person med en god tilstand af immunitet, forekommer disse sygdomme som regel ikke eller fortsætter i let.
Ifølge denne klassifikation er primære immunodeficies opdelt i 5 grupper:
- Usufficiens of humoral immunitet
- Usufficiens af cellulær immunitet
- Kombineret utilstrækkelig til humoral og celleimmunitet
- Foocytmangel
- Manglende komplement
Grundlaget for den moderne klassificering af primær immundefekt er det præference nederlag af et eller andet immunitetsniveau.
Fogocytosis mangel
Fagocitmangel er 10-15% af al primære immundefekt. Phagocytmangel skyldes nedsat proliferation (vækst), differentiering, kemotaxis (cellulær cellereaktion som reaktion på effekten af kemikalier) neutrofiler og makrofager - celler af aktivt involveret i fagocytose og faktisk overtræder phagocytose-processen.
Phagocytiske celler (celler, der deltager i fagocytose), er repræsenteret ved leukocytter og celler af makrofager udfører en vigtig rolle i beskyttelsen af kroppen mod glasset bakterier og andre intracellulære mikroorganismer. Den udtalte utilstrækkelige polymorfe leukocytter (neutropeni - et fald i antallet af neutrofiler i den generelle blodprøve) kan føre til udvikling af en massiv bakteriel infektion.
Af særlig betydning er to genetiske defekter, der forstyrrer fagocytternes funktion, med hvilken forekomsten af alvorlige sygdomme er forbundet, ofte med død - kronisk granulomatose (hvis årsag består i strid med iltreduktionsmekanismen) og manglen på vedhæftning ( adhæsion) af leukocytter (på grund af defekter af gener).
Utilsigtet komplementsystem
 Manglen på komplementsystemet er ikke mere end 2% af al primær immundefekt, manifesteres af en lidelse af opsoniseringen (processen med interaktion med bakterier), fagocytose (processen med at fange og fordøje mikroorganismer) og ødelæggelsen af mikroorganismer og er ledsaget af alvorlige infektioner, op til udviklingen af sepsis. Komplementmangel overholdes ofte i autoimmune sygdomme, for eksempel med en systemisk rød loll.
Manglen på komplementsystemet er ikke mere end 2% af al primær immundefekt, manifesteres af en lidelse af opsoniseringen (processen med interaktion med bakterier), fagocytose (processen med at fange og fordøje mikroorganismer) og ødelæggelsen af mikroorganismer og er ledsaget af alvorlige infektioner, op til udviklingen af sepsis. Komplementmangel overholdes ofte i autoimmune sygdomme, for eksempel med en systemisk rød loll.
Manglen på komponenterne i den klassiske vej af komplementaktiveringen forårsager en forudsætning for sygdomme som følge af lidelser i dannelsen af immunkomplekser, for eksempel til fremkomsten af systemisk rød lupus. Komplementunderskud fører til en stigning i kroppens følsomhed over for purulente infektioner. Usufficiens af terminalkomponenterne i komplementsystemet samt komponenter i en alternativ vej skaber en særlig prædisponering til infektioner - gonoré og meningitis. Dette angiver tydeligt en vigtig rolle som en alternativ måde for at aktivere komplementet og den ødelæggende membran af komplekset i processen med at fjerne bakterierne af de specificerede arter.
De mest alvorlige lidelser i komplementfunktionerne, der er forbundet med injektionen af komponenterne i komplementsystemet, hvilket fører til et arvigt angriefsedemning (en allergisk reaktion, ledsaget af et ødem af slimhinden af mundhulen og øvre luftveje); Sygdommen overføres som et autosomalt dominerende tegn. Komplementsysteminhibitoren blokerer ikke kun den klassiske integrationsvej for komplementet, men undertrykker også aktiviteten af blodkoagulationssystemet. Dybest set er form af utilstrækkelighed af komponenter af komplementet arvet som autosomale recessive tegn.