Gentons sygdom - Covaric Ailment. Han er til stede i det menneskelige genom, så mener du en sygdom forudbestemt. Men gentons sygdom manifesteres i voksenalderen. Og bogstaveligt talt ødelægger livet af både patienten selv og hans kære, måske indtil for nylig og ikke gættet «arv».
Indhold
 Ligegyldigt hvor trist det er at genkende, men der er ikke perfekte mennesker. Ingen af os kan desværre ikke «prale» Manglen på ubehagelige mutationer i sit genom - den arvelige fragt modtaget fra mange hundrede generationer af forfædre, løgnen. Kun nu mutationer er forskellige: en (som dem, der er ansvarlige for udviklingen af hæmofili eller fibrose) omtrent uden unødvendig «Ceremonier» fratage en person mulighed for fuldt udviklet liv fra fødslen, andre - kun «løfte» nogensinde «udøse» I tidlig aterosklerose, allergiske sygdomme eller maligne tumorer.
Ligegyldigt hvor trist det er at genkende, men der er ikke perfekte mennesker. Ingen af os kan desværre ikke «prale» Manglen på ubehagelige mutationer i sit genom - den arvelige fragt modtaget fra mange hundrede generationer af forfædre, løgnen. Kun nu mutationer er forskellige: en (som dem, der er ansvarlige for udviklingen af hæmofili eller fibrose) omtrent uden unødvendig «Ceremonier» fratage en person mulighed for fuldt udviklet liv fra fødslen, andre - kun «løfte» nogensinde «udøse» I tidlig aterosklerose, allergiske sygdomme eller maligne tumorer. i øvrigt «Løfte» måske «hold ikke nede» - Hvis en transportpersons livsstil ikke vil give af denne grund. Men der er mutationer med en fundamentalt forskellig effekt: de erklærer absolut sig selv, men de vælger det mest uhensigtsmæssige øjeblik - som Pushkin Silvio fra «Skud». Det er svært at finde i naturen noget mere grusomt og uretfærdigt end dødelig arvelig sygdom med en forsinket manifest.
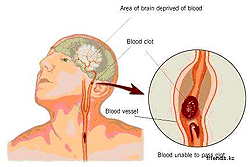
I 1872 beskrev den amerikanske læge George Hotthtton først detaljeret symptomerne på alvorlig sygdom, kendetegnet ved familiekarakter. Alle patienter (både mænd og kvinder) var generelt sunde mennesker før… Moden alder. I perioden mellem 30 og 50 år begyndte de nervesystemets progressive og irreversible patologi - svagt op til det fulde tab af selvbetjeningskompetencer og øget diskordineret motoraktivitet - konvulsiv sammentrækning af ansigtets muskler, lemmer. Gait lignede således med stærk forgiftning og skarpe ukontrollable bevægelser af patienternes hænder og fødder (t.N. HYHETISKE HYPERCINES) Modtaget et navn i medicin «Dans af St. Vitta».
Sygdommen blev kaldt Hantington's Chorees - og allerede i begyndelsen af det tyvende århundredes medicinske videnskab var det klart, at sådanne havde en arvelig karakter: chancerne for at vise eller ikke vise de samme symptomer i voksenalderen for patientens børn er 50x50.
Huntingtons Chorea er desværre ikke så sjældent, som det vil, - kun i USA, mere end 30.000 mennesker lider i dag, og omkring 150.000 er i risikogruppen.
Tragedien af Genthatons sygdom er ikke kun, at det for trangende bryder menneskelivet i den mest frugtbare periode af hendes periode, men også at patientens efterkommere er tvunget til at leve med en konstant tanke, at den samme skæbne måske falder i tide og deres. Såvel som deres børn, børnebørn, børnebørn… Og måske - og nej. Dommer kan kun tid. Det er overflødigt at sige, hvad der er «Divination» helt bidrager ikke til sindsroen af eksistensen og opførelsen af langsigtede livsplaner. Situationen forværres af, at denne sygdom er uhelbredelig til denne dag.
Det skete så i 80'erne, Korea Huntington kom ind i synet af molekylær genetik. En væsentlig rolle i dette blev spillet af, at forskerne havde prøver af DNA-medlemmer af meget store familier, hvor Kharya Genthoton blev overført gennem mange generationer. For eksempel, i en familie fra Iowa 22 af 41 medlemmer ledet af denne sygdom. Og den venezuelanske familie, der bor på kysten af Lake Maracaibo, omfattede 250 (!) Patienter med Chorees og 727 personer i en gruppe på 50% risiko. Alle var efterkommere af en portugisisk sømand - en bærer af en mutation - bosatte sig i disse kanter i 1800. En sådan stor mængde biologisk materiale gjorde det muligt at genetisk kortlægning (definition af placering) gen, der er ansvarlig for Hornington's Chore. Hvad blev med succes gennemført i maj 1983.
Til rådighed for medicin faldt en enestående mulighed - den nøjagtige diagnose af Khalei Genretton ophørte med at have brug for kliniske tegn, og dermed - og i en alder af enkeltpersoner. En analyse af DNA'et fra den unge mand eller en pige, en enårig baby eller endda en 10-ugers embryo tilladt at sige med tillid: Denne person vil lide ovennævnte chorea, og denne - nej.
Presymptomatisk diagnostik - så begyndte at kalde det - det viste sig at være et vanskeligt problem for bioethik. Er det nødvendigt at kende barnet, at til hans rådighed ikke er alt liv, men kun 35-40-50 år? Har du brug for at vide nøjagtigt om din genetiske fatumen voksen søn eller datter syg choree gentonon? Eller det er bedre at forblive i ængstelig uvidenhed - fordi sidstnævnte giver i det mindste noget håb, mens passagen af molekylær genetisk test vil implementere alle punkter over «JEG». Hver person reagerer på nyheden om sådan dramatisk mætning på forskellige måder - det er lettere at leve «Under lamoklovy sværd». Resultaterne af præimptomatisk testning har gentagne gange fungeret som en grund til selvmordsforsøg af de undersøgte.
Et andet spørgsmål: Skal de vide om den nøjagtige genetiske status for mennesker fra risikogruppen af deres kone eller mand? Og endelig en endnu vanskeligere situation: Hvis en person fra en risikogruppe, der ikke ønsker at gennemgå en undersøgelse, er planer om at konfigurere - er det nødvendigt at undersøge embryoet? Og om nødvendigt - så hvem skal vide om resultaterne af denne undersøgelse? Når alt kommer til alt, hvis embryoet er en bærer af dødelig mutation, kan hans fødsel forhindres. Men det vil automatisk rydde op på hans forælders status - men det er måske ikke, at du ikke vil vide om denne status. Et andet aspekt af problemet med oplysninger om en persons genetiske status fra risikogruppen - om det er nødvendigt at levere sådanne forsikringsselskaber, arbejde og låne? Generelt, endnu en gang, skitseret tid - svarene på alle disse spørgsmål er ikke let at finde i religiøse øvelser og universelle moralsystemer, der eksisterer i dag.